About This Project
Cancer remains one of the most challenging health issues worldwide. Current therapies like chemotherapy, radiation, and surgery often come with significant side effects (off-target effects) and limited effectiveness. To address these limitations, my project proposes leveraging synthetic genetic pathways for cancer therapy. This project capitalizes on the processes involved in DNA damage response to create a targeted and low-cytotoxicity approach to combat cancer.
Ask the Scientists
Join The DiscussionWhat is the context of this research?
We've put together a small but mighty team of scientists to try and come up with a novel solution to cancer. We've devised a theoretical detection method and response mechanism which we believe will be able to detect and induce Apoptosis (programmed cell death) in cells carrying DNA mutations. We call this new possible treatment: GC (or genetic circuit) Therapy. Using natural cell signaling mechanisms we believe its possible that this project could mean a breakthrough in cancer research.
Where will this research take place?

We've partnered with the Non-profit BUGSS lab in Baltimore to do initial research involving the creation of the genetic circuit itself, and testing in cells.
What is the significance of this project?
Firstly, our treatment has the potential to work on a large variety of cancer types, from skin to brain. The therapy works by using a complex detection mechanism which can receive signals from a variety of cancer mutations. Secondly, the genetic circuit detects existing signals that would normally be sent in cells in response to mutations. This enables the treatment to pick up on cancer cells' abilities to detect themselves. And lastly, the key is the detection mechanism built into the genetic circuit. With what we designed, we can pick and choose what downstream gene is activated to result in different results based on weather a mutation is detected.
What are the goals of the project?
The primary objective is to develop a synthetic genetic circuit capable of selectively inducing apoptosis in cancerous cells while sparing healthy ones. However, we believe that the circuit could be altered to perform a multitude of tasks (e.g., DNA repair, and cellular senescence) in response to DNA damage detection. Key goals of the project include (1) engineering a genetic circuit comprising two main operons: MDM2 and Complex-9, designed to exploit cellular response mechanisms to DNA damage. (2) validating the effectiveness and specificity of the circuit in targeting mutated cells in vitro. And lastly, (3) investigating potential modifications and applications of the circuit for different types of cancer and therapeutic interventions.
Budget
Materials involved with plasmid construction, cell culture, and cell transfection are estimated below. Total costs may vary depending on the availability of equipment and reagents. I anticipate that plasmid construction, and cell culture experiments will be performed at Baltimore Underground Science Space (BUGSS), but (phase 2) animal models and more complicated tissue cultures with other cell lines will be performed at another institution to be determined.
Endorsed by

 Project Timeline
Project Timeline
The Initial research timeline (excluding in vivo models) is predicted to take around 4 months in total. This includes time to construct the Genetic Circuit, package it in lipid-based nanoparticles and hydrogel, and test it on the cancer cell lines. We plan on sharing our all of our experimental results with our funders by giving access to data results and regularly posting to our lab notebook.
May 09, 2024
Project Launched
Jun 01, 2024
Building The Genetic Circuit/Construct
Jul 01, 2024
Packaging GC Therapy
Aug 31, 2024
Testing Treatment On Cell Lines
Oct 31, 2024
Writing Manuscript + Sharing Results To Funders
Meet the Team


Affiliates
Team Bio
Our team, lead by Dylan Paoletti and mentored by Dr. Peter Evans boasts a potent blend of experience and innovation. With the sharp acumen and perspective of a high school student, the seasoned experience of some of the most brilliant members of the lab (Dr. Evans), we're uniquely positioned to pioneer advancements through a combination of ideas and action. We are an interdisciplinary team with various lab skills and are committed to accomplishing our objectives.
Dylan Paoletti
Dylan is a student who prolifically comes up with new ideas to try and solve problems with science and is currently trying to tackle cancer.
Peter Evans
Dr. Evans is a molecular and synthetic biologist. He has degrees in public health and molecular biology from Johns Hopkins, University of Wisconsin, and University of Connecticut, and has been working in these fields since the early 90's. Linkedin
Additional Information
Scientific Reasoning
The underlying principles of the proposed genetic circuit for cancer therapy are rooted in the interplay between TP53 (p53) and MDM2, and how this interaction is regulated by various outside proteins to prevent the degradation of p53 by ubiquitination in response to DNA damage (Ref. The MDM2-p53 pathway revisited).

TP53, commonly referred to as p53, is a tumor suppressor protein that plays a pivotal role in regulating cell growth and preventing cancer development. It acts as a transcription factor, activating genes involved in cell cycle arrest, DNA repair, and apoptosis in response to cellular stressors like DNA damage or oncogene activation.
MDM2 is a negative regulator of p53. It functions primarily as an E3 ubiquitin ligase, targeting p53 for ubiquitination and subsequent proteasomal degradation. Under normal conditions, MDM2 tightly regulates p53 levels, maintaining them at a basal level. This regulation is crucial for preventing excessive p53 activity, which could lead to cell cycle arrest or apoptosis unnecessarily.
The interaction between p53 and MDM2 is tightly regulated and subject to modulation by various cellular proteins. The proteins ARF, ATM, ATR, CHK1, and CHK2 interact with p53 and MDM2 to prevent p53 denaturation in response to DNA mutations through various mechanisms:
ARF (ADP ribosylation factor): ARF induces p53 accumulation by binding to the MDM2 acidic domain, inhibiting p53 ubiquitination, and promoting p53 stabilization.
ATM (Ataxia Telangiectasia Mutated): ATM phosphorylates MDM2 upon activation, inhibiting MDM2's ability to poly-ubiquitinate p53. This phosphorylation prevents p53 degradation by MDM2, leading to p53 stabilization and protection from denaturation.
ATR (ATM and Rad3-related): ATR phosphorylates MDM2 and MDMX in response to DNA damage, reducing their affinity for p53 and favoring p53 activation. This phosphorylation of negative regulators of p53 leads to their destabilization and degradation, contributing to p53 stabilization and preventing denaturation.
CHK1 (Checkpoint Kinase 1): CHK1 is rapidly phosphorylated by active ATR in response to DNA damage, leading to its full activation. CHK1 phosphorylates and inactivates proteins like Cdc25A and Cdc25C, contributing to cell cycle arrest and preventing DNA synthesis. The activation of CHK1 is part of a complex network that stabilizes p53 and prevents its denaturation in response to cellular stresses.
CHK2 (Checkpoint Kinase 2): CHK2 is activated by ATM in response to DNA damage and plays a role in regulating the cell cycle and DNA repair processes. It contributes to the stabilization of p53 through a sophisticated network of interactions that prevent denaturation and ensure proper cellular responses to mutations.
Essentially, a various array of DNA damage detection proteins interact with MDM2-p53 to stop p53 denaturing in cancerous cells. This is key to the overall idea.

Cellular Response Mechanisms:
The genetic circuit capitalizes on cellular response mechanisms to DNA damage, particularly involving proteins that are involved in the MDM2-p53 signaling pathways in mutated cells. An iCasp-9 and p53 fusion protein (which I’ve named Complex-9), will be ubiquinated and destroyed in normal cells. However, in cancerous cells, the protein won't be destroyed, leading to cell death.
The genetic circuit created is composed of two main operons. The MDM2 operon which is responsible for Complex-9 degradation through ubiquitination starting at the p53 portion, enables the destruction of the iCasp-9 portion of the fusion protein, and the Complex-9 operon which encodes the production of the Complex-9 fusion protein. The MDM2 gene is regulated under strong constitutive expression of the CMV promoter. The Complex-9 is under the control of the PGK promoter (a promoter of medium-low strength). Due to the two operons' close proximity and the greater strength of the CMV promoter, the MDM2 will be able to fully prevent protein expression of Complex-9 in non-cancerous cells. However, in cancerous cells featuring mutagenesis, a variety of proteins are able to detect DNA damage and respond accordingly. The most notable and understood of which include those earlier mentioned (ARF, ATM, ATR, ChK1, ChK2). For this reason, the circuit designed contains a fusion protein (complex-9) which also encodes an executioner Caspase; this enables apoptotic function only in mutated cells containing the plasmid. Unlike other treatment options (such as: chemotherapy, radiation, surgery), synthetic genetic pathways enable distinct, low cytotoxicity options that target mutated cells indiscriminately but with low off-target effects.
Hypothesis:
Leveraging the connection between TP53 and MDM2, we hypothesize that this circuit will effectively target mutated cells, aided by differential promoter strengths (CMV for MDM2, PGK for Complex-9), and DNA damage signaling proteins (ARF, ATM, ATR, ChK1, and ChK2). We anticipate reduced off-target effects compared to conventional therapies, offering a promising strategy for cancer treatment.

(NOTE: A fusion protein consisting of p53 and an executioner caspase[iCasp-9] is to be named the “Complex-9” protein to avoid confusion.
Additionally, the figure above labels the protein “DNA Damage Response Protein”; this characterizes proteins such as ARF, ATM, ATR, ChK1, and ChK2.)
Approach
Genetic Circuit Design:
The core of my approach lies in the design of a genetic circuit consisting of two main operons (as well as a simple GFP marker operon):
Complex-9 Operon: Controlled by the PGK promoter, this operon encodes the fusion protein Complex-9, comprising p53 linked to iCasp-9. Studies comparing mammalian promoter strength have shown that the PGK promoter is significantly weaker than the CMV promoter, and it’s why it’ll be used for this study. The degradation of Complex-9 by MDM2 is exploited to induce apoptosis selectively in cancerous cells, and it’s why Complex-9 is under a promoter of weaker strength.
MDM2 Operon: Regulated by the strong CMV promoter, this operon facilitates the degradation of the iCasp-9 portion of Complex-9. The MDM2 gene prevents the expression of Complex-9 in non-cancerous cells, thus minimizing off-target effects.

In between the p53 and IRES, a sequence consisting of glycine linker followed by the iCasp-9 ORF will be incorporated (after removal of the p53 stop codon). Additionally, directly downstream of the CMV promoter the MDM2 ORF followed by an SV40 poly A signal will be inserted.
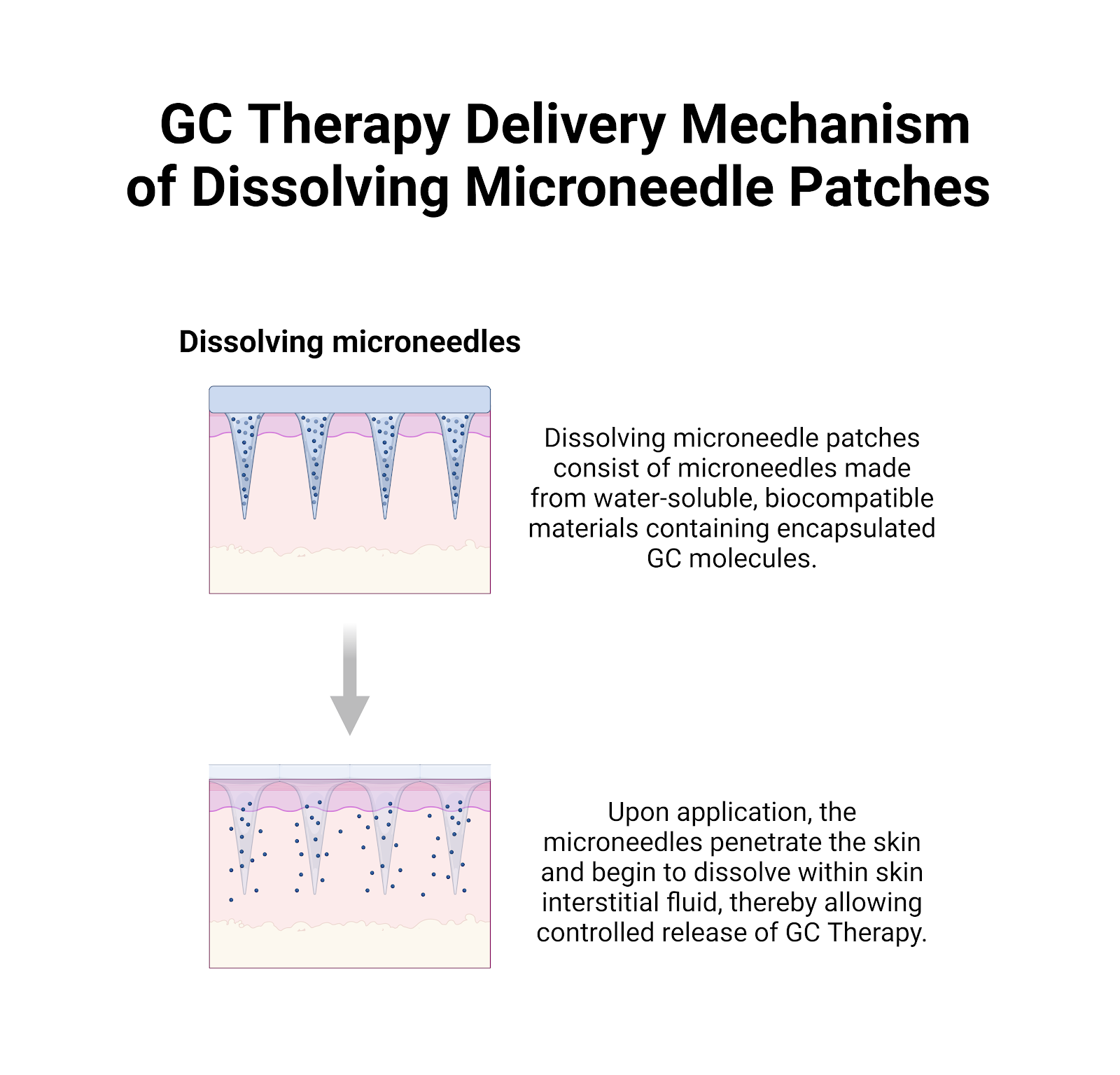
Delivery Method:
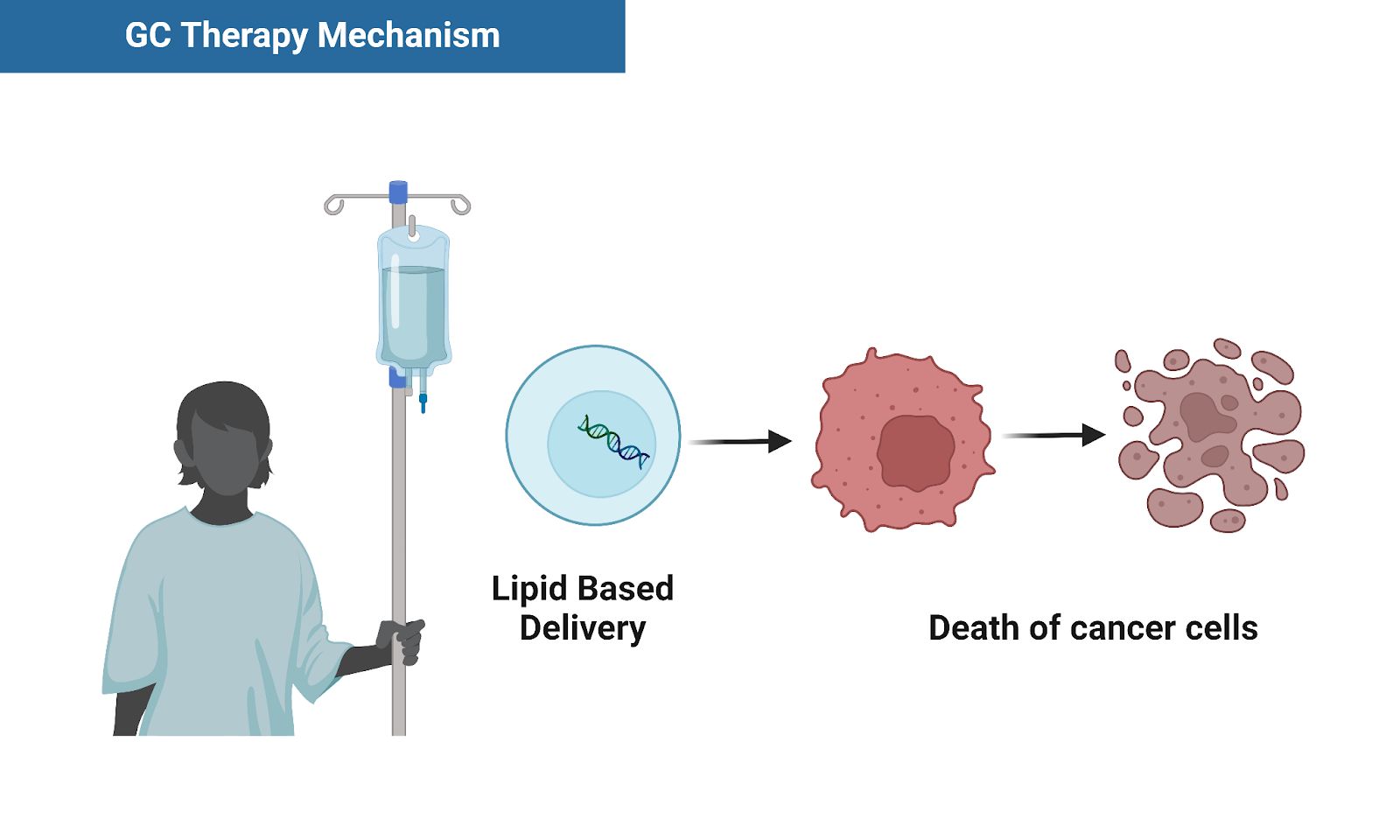
We're using hydrogel based microneedle patches to deliver lipid particles containing GC Therapy.

Components of Genetic Circuit:
CMV Promoter | A cytomegalovirus promoter commonly used in molecular biology to drive strong gene expression in mammalian cells. |
PGK Promoter | Phosphoglycerate kinase promoter, another promoter commonly used to drive medium-low expression in mammalian cells. |
Glycine Linker | A short amino acid sequence (usually consisting of glycine residues) is used to join protein ORFs, offering dual expression and flexibility. |
Complex-9 (p53-iCasp9 fusion protein) | Fusion protein combining the tumor suppressor 53 (TP53) ORF along with the iCasp-9 ORF for inducible apoptosis. |
IRES | Internal Ribosome Entry Site- allows for the bicistronic expression of genes under the same promoter. In my circuit, this includes complex-9 and EGFP under the PGK promoter. |
SV40 poly A | The polyA signal from the simian virus 40 (SV40), used to terminate transcription in mammalian expression systems. |
AmpR Promoter | A promoter sequence associated with the ampicillin resistance used in plasmids for bacterial selection and cloning. |
Kan/NeoR | Antibiotic resistance markers used to select for bacteria that have taken up the plasmid containing the genetic circuit of interest. |
F1 ori | Origin of replication used in plasmids for propagating the plasmid of interest in bacterial expression systems. |
ColE1 ori | Origin of replication from the ColE1 plasmid, it’s commonly used in molecular cloning to propagate plasmids in E. coli. |
MDM2 | Protein that regulates the activity of p53 by ubiquitination. It plays an important role in making my genetic circuit inducible. |
Experimental Details
Cell Culture:
Through In vitro experiments using the biosafety level (BSL)1 cancer cell line A-431 and the HEKa cell line I will validate the functionality and specificity of the genetic circuit with electrophoresis to detect DNA fragmentation (a hallmark of apoptosis). Optimization strategies will involve fine-tuning promoter strengths (which may include considering alternative promoters for the circuit and/or inducing PCR mutagenesis depending on the initial result(s), and possibly exploring alternative downstream ORFs within the feedback loop to enhance efficacy and versatility.
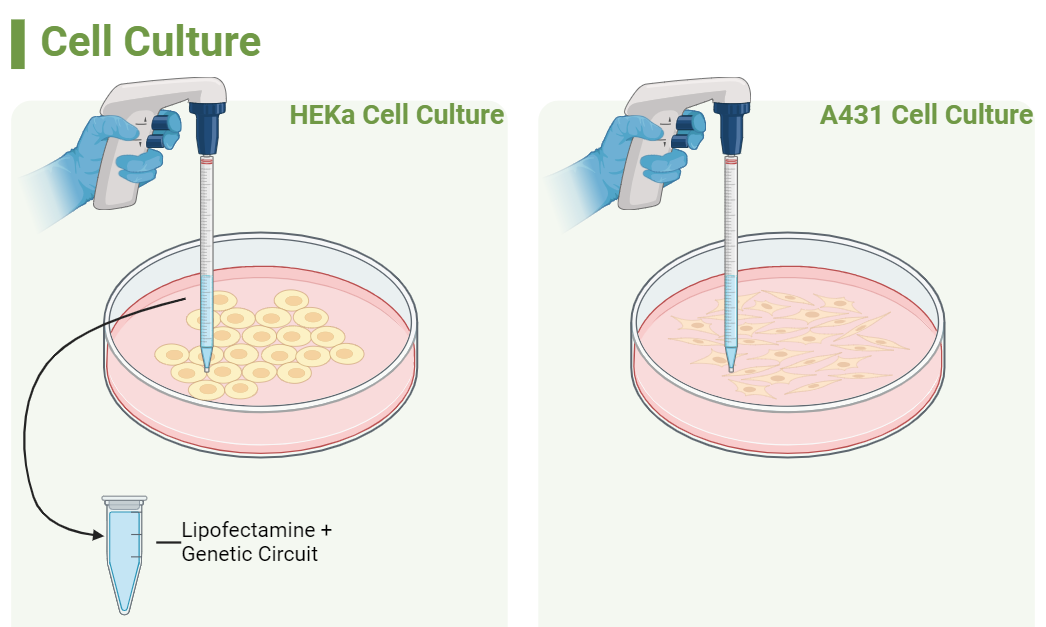
Cell Models: These two cell lines will be used to compare the genetic circuit in both cancerous and non-cancerous cells
A-431: The A431 cell line, derived from human skin carcinoma, exhibits elevated expression levels of epidermal growth factor receptor (EGFR). Among tumor suppressor genes, p53 is notably the most commonly mutated across all cancer types. In the case of the A-431 cell line, it carries two missense mutations located in exon 5 of the p53 gene, resulting in a reduced expression of p53 mRNA to only 50% of the normal level.
HEKa: HEKa cells, or human epidermal keratinocytes, are derived from human skin tissue and are widely used in research related to dermatology, wound healing, and skin biology.
Cell Transformation
In my project, I propose to utilize Lipofectamine as the delivery method for introducing the genetic circuit into both cell types. Lipofectamine has demonstrated high efficiency in transfecting various cell types, making it an ideal choice for the study.
Transformed Cell Characterization
An agarose gel will be run to detect DNA fragmentation (a late stage symptom of Apoptosis) in both cancerous and non-cancerous cells which receive the treatment. The genetic circuit contains a GFP marker, so transformation can be verified because all transformed cells should express a green fluorescence under UV light. Additional testing involving fluorescence microscopy Assays (like the TUNEL Assay), and flow cytometry Assays (like the Annexin V Assay) could also potentially be done at an institution to be determined with the required instruments.
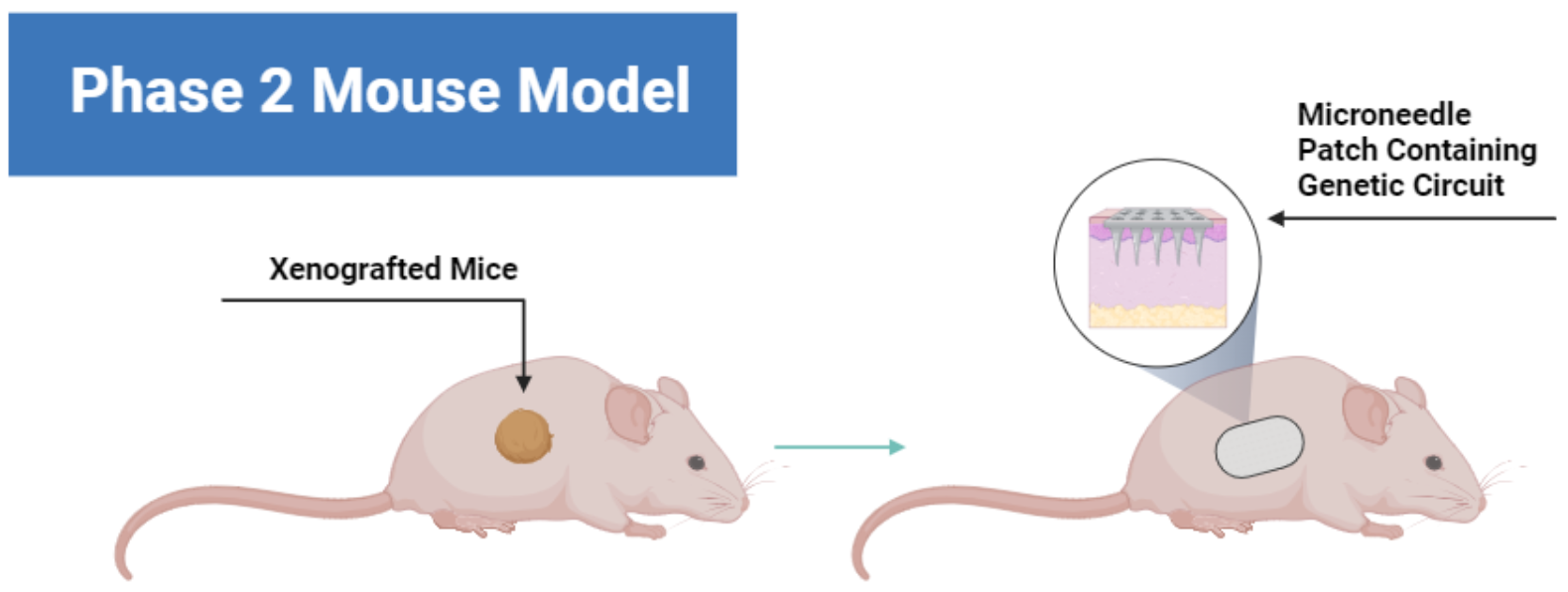
Future goals (Phase 2):
Additionally, I’d like to think forward to the prospect of creating or utilizing a skin cancer model that imitates invasive skin cancer. This can be done using co-cultures or a xenografted mouse model.
Co-cultures:
The figure below is a demonstration of such a culture where a microneedle needle patch containing the genetic circuit is used to deliver it in the cell model.
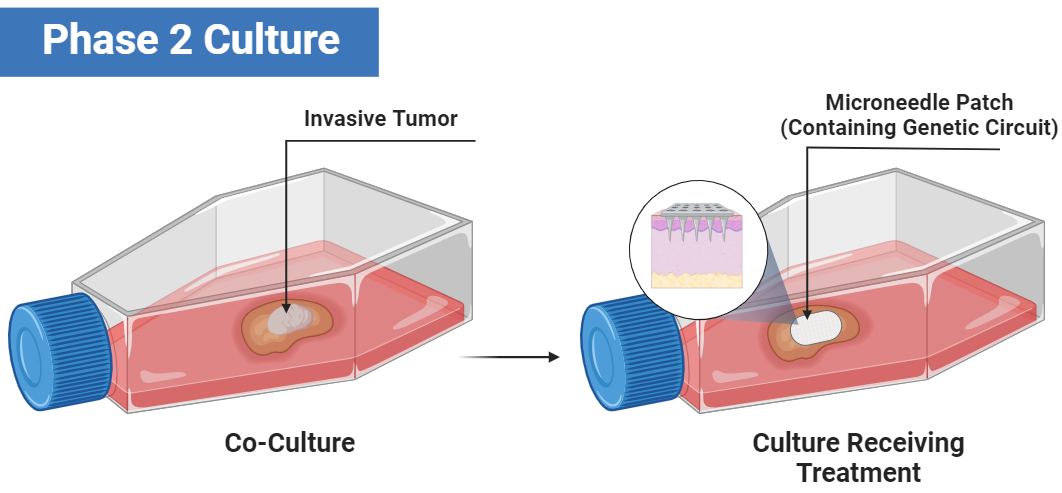
In Vivo Testing:
Similar to the figure above, this illustration depicts further testing. I propose the utilization of xenografted mice models, where human cancer cells are transplanted into mice. Below is a figure showcasing the testing of the same delivery in a mouse as the previous figure but in a mouse.
Budget:
Materials involved with plasmid construction, cell culture, and cell transfection are estimated below. Total costs may vary depending on the availability of equipment and reagents. I anticipate that plasmid construction will be performed at Baltimore Underground Science Space (BUGSS), and cell culture experiments (and phase 2) will be performed at another institution to be determined.
References
Abcam. (2019, July 18). Apoptosis Assay and Apoptosis Marker Guide | Abcam. Abcam.com. https://www.abcam.com/kits/apo...
Abuetabh, Y., Wu, H. H., Chai, C., Al Yousef, H., Persad, S., Sergi, C. M., & Leng, R. (2022). DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Experimental & Molecular Medicine. https://doi.org/10.1038/s12276...
Ashcroft, M., Kubbutat, M. H. G., & Vousden, K. H. (1999). Regulation of p53 Function and Stability by Phosphorylation. Molecular and Cellular Biology, 19(3), 1751–1758. https://www.ncbi.nlm.nih.gov/p...
Cheng, Q., & Chen, J. (2010). Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle (Georgetown, Tex.), 9(3), 472–478. https://www.ncbi.nlm.nih.gov/p...
Hu, W., Feng, Z., & Levine, A. J. (2012). The Regulation of Multiple p53 Stress Responses is Mediated through MDM2. Genes & Cancer, 3(3-4), 199–208. https://doi.org/10.1177/194760...
Lavin, M. F., & Gueven, N. (2006). The complexity of p53 stabilization and activation. Cell Death & Differentiation, 13(6), 941–950. https://doi.org/10.1038/sj.cdd...
Levine, A. J. (2019). The many faces of p53: something for everyone. Journal of Molecular Cell Biology, 11(7), 524–530. https://doi.org/10.1093/jmcb/m...
Liu, Y., Tavana, O., & Gu, W. (2019). p53 modifications: exquisite decorations of the powerful guardian. Journal of Molecular Cell Biology, 11(7), 564–577. https://doi.org/10.1093/jmcb/m...
Matassov, D., Kagan, T., Leblanc, J., Sikorska, M., & Zakeri, Z. (2004). Measurement of apoptosis by DNA fragmentation. Methods in Molecular Biology (Clifton, N.J.), 282, 1–17. https://doi.org/10.1385/1-5925...
Nag, S., Qin, J., Srivenugopal, K. S., Wang, M., & Zhang, R. (2013). The MDM2-p53 pathway revisited. Journal of Biomedical Research. https://doi.org/10.7555/jbr.27...
Norris, P. S., & Haas, M. (1997). A fluorescent p53GFP fusion protein facilitates its detection in mammalian cells while retaining the properties of wild-type p53. Oncogene, 15(18), 2241–2247. https://doi.org/10.1038/sj.onc...
Protein P53 - an overview | ScienceDirect Topics. (n.d.). Www.sciencedirect.com. https://www.sciencedirect.com/...
Rocha, S., Garrett, M. D., Campbell, K. J., Schumm, K., & Perkins, N. D. (2005). Regulation of NF-κB and p53 through activation of ATR and Chk1 by the ARF tumour suppressor. The Embo Journal, 24(6), 1157–1169. https://doi.org/10.1038/sj.emb...
Vogelstein, B. (2010). p53, Cancer | Learn Science at Scitable. Nature.com. https://www.nature.com/scitabl..
Project Backers
- 24Backers
- 34%Funded
- $2,362Total Donations
- $98.42Average Donation